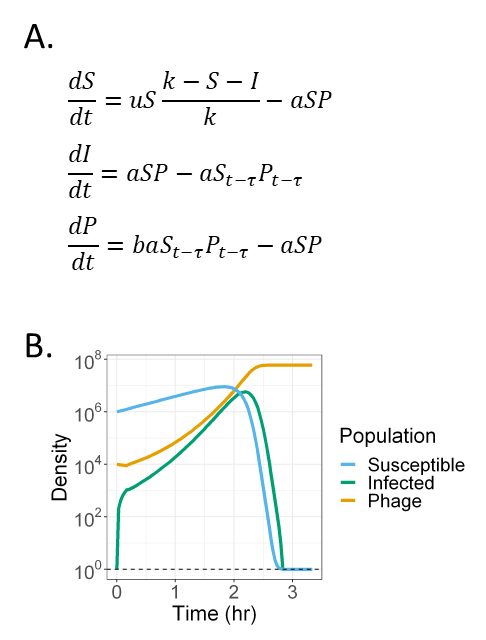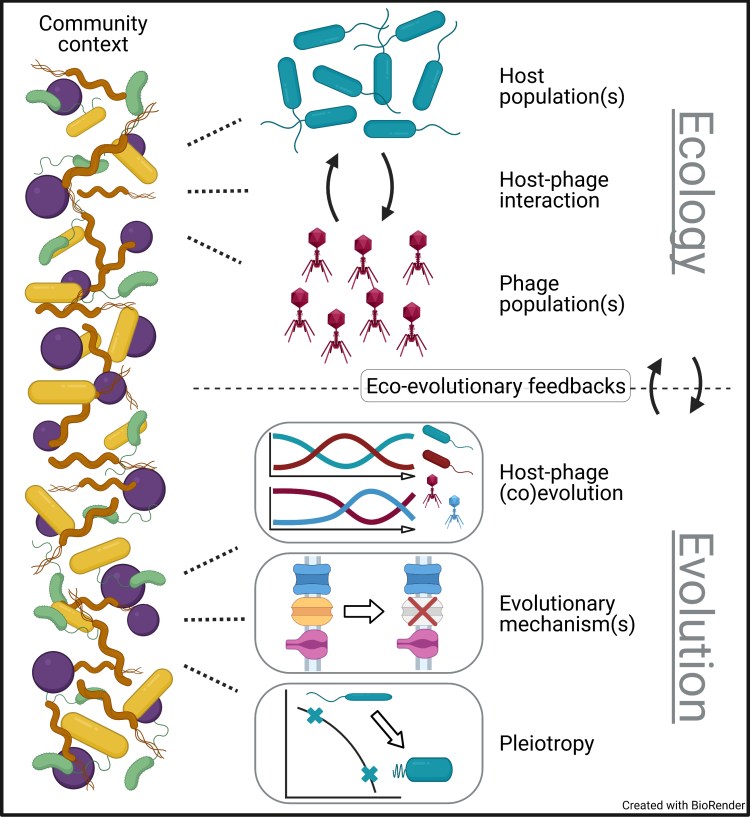
Phage-bacteria coevolution in multispecies communities
Bacteria-phage symbioses are ubiquitous in nature and serve as valuable biological models. Historically, the ecology and evolution of bacteria-phage systems have been studied in either very simple or very complex communities. Although both approaches provide insight, their shortcomings limit our understanding of bacteria and phages in multispecies contexts. To address this gap, we recently published a review and meta-analysis arguing that non-host bacterial species are an important driver of phage-bacteria ecology and evolution:
M Blazanin and PE Turner. “Community context matters for bacteria-phage ecology and evolution.”
I am also currently working on a paper where we test these sorts of ideas in a model in vitro cystic fibrosis bacterial community I developed during my PhD.
gcplyr: an R package for microbial growth curve data analysis
Characterization of microbial growth is of both fundamental and applied interest. Modern platforms can automate collection of high-throughput microbial growth curves, necessitating the development of computational tools to handle and analyze these data to produce insights. However, existing tools are limited. Many use parametric analyses that require mathematical assumptions about the microbial growth characteristics. Those that use non-parametric or model-free analyses often can only quantify a few traits of interest, and none are capable of importing and reshaping all known growth curve data formats. To address this gap, I developed and recently released a new R package:
M Blazanin. “gcplyr: an R package for microbial growth curve data analysis.”
gcplyr can flexibly import growth curve data in every known format, and reshape it under a flexible and extendable framework so that users can design custom analyses or plot data with popular visualization packages. gcplyr can also incorporate metadata and generate or import experimental designs to merge with data. Finally, gcplyr carries out model-free and non-parametric analyses, extracting a broad range of clinically and ecologically important traits.

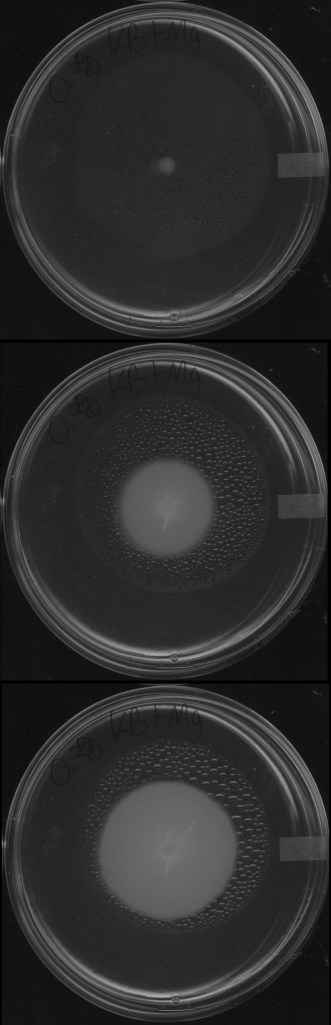
Parasite-driven evolution of bacterial avoidance
In the face of ubiquitous threats from parasites, hosts often evolve strategies to resist infection or to altogether avoid contact with parasites. At the microbial scale, bacteria frequently encounter viral parasites, bacteriophages. While bacteria are known to utilize a number of strategies to resist infection by phages, and can physically navigate their environment using complex motility behaviors, it is unknown whether bacteria evolve avoidance of phages. In order to answer this question, we combined experimental evolution and mathematical modeling to test whether, and under what conditions, bacteria will evolve to avoid viral parasites.
Growth curves as a high-throughput method to quantify phage-bacteria interactions
Quantitative measures of bacteriophage infectivity are essential to understanding bacteria – phage interactions across a range of domains and applications, from human health to fundamental research. However, existing methods, like cross-streaks, spot tests, and efficiency of plaquing assays, have limitations. Most notably, these assays experience strong tradeoffs between the resolution of infectivity data and the throughput of the assay, with high-throughput assays providing little more than a binary measure of infectivity. We set out to bridge this gap by improving a high-throughput method already in widespread use: growth curves. Growth curves consist of a time series of bacterial density data, and are often used to identify the presence of phage-induced lysis. In this project, we use mathematical modeling and in vitro experiments to show whether and how growth curve data can, in fact, produce high-resolution measures of phage infectivity.